






Built for the Needs of Your Industry
Be confident you have all the information needed for variant interpretation ACMG secondary findings, cardiac panel, hereditary cancer panel, curated gene & variant level information
Reduce Turnaround Time
Explore our continually growing catalog of pre-classified variants, designed to facilitate the rapid uncovering and interpretation of novel genetic variations.
Access valuable gene and variant summary information to streamline your analysis and deepen your insights.

Increase Diagnostic Yield
Gain confidence in identifying more disease-causing variants leveraging our Genomic Intelligence Platform.
Our genomic language processing technology empowers you to uncover more disease-causing variants with precision.

Accelerate Throughput
Optimize your workflows by rapidly prioritizing variants of disease-causing genes.
This efficient process reduces resources, simplifying your analysis and enabling a more targeted approach.
The Mastermind Genomic Intelligence Platform
Mastermind gives you access to all the information that's available on your disease of interest already curated and ready to go.
You can search by variant, gene, disease, therapy, and more. Mastermind contains the full text of more than 9.5 million publications, which are easily searchable by gene, variant, disease, and other key terms.
Save Time and Minimize Expenses
Expedite your research workflow with instant access to the entirety of genomic research, pre-annotated by gene, variant, disease, and therapeutic.
Accelerate Trial Eligibility Decisions
Get the information you need to make an educated decision faster through an easy-to-use search interface that returns prioritized, filterable literature results for identified variants.
Characterize and Prioritize Targets and Programs
Accelerate research on potential targets and programs through prioritized and filterable literature results for the gene and/or disease of interest.
Thousands of labs are using Mastermind to increase diagnostic yield.
Start searching the world's most comprehensive source of genomic evidence. Create your free account today.
Mastermind PRO offers users unparalleled insights at gene and variant level from over 10M full text articles, over 3.7M supplemental text and growing daily.
Through our powerful combination of genomics-focused AI and expert human curation, Mastermind offers a level of depth and precision that elevates variant interpretation for researchers and clinicians.
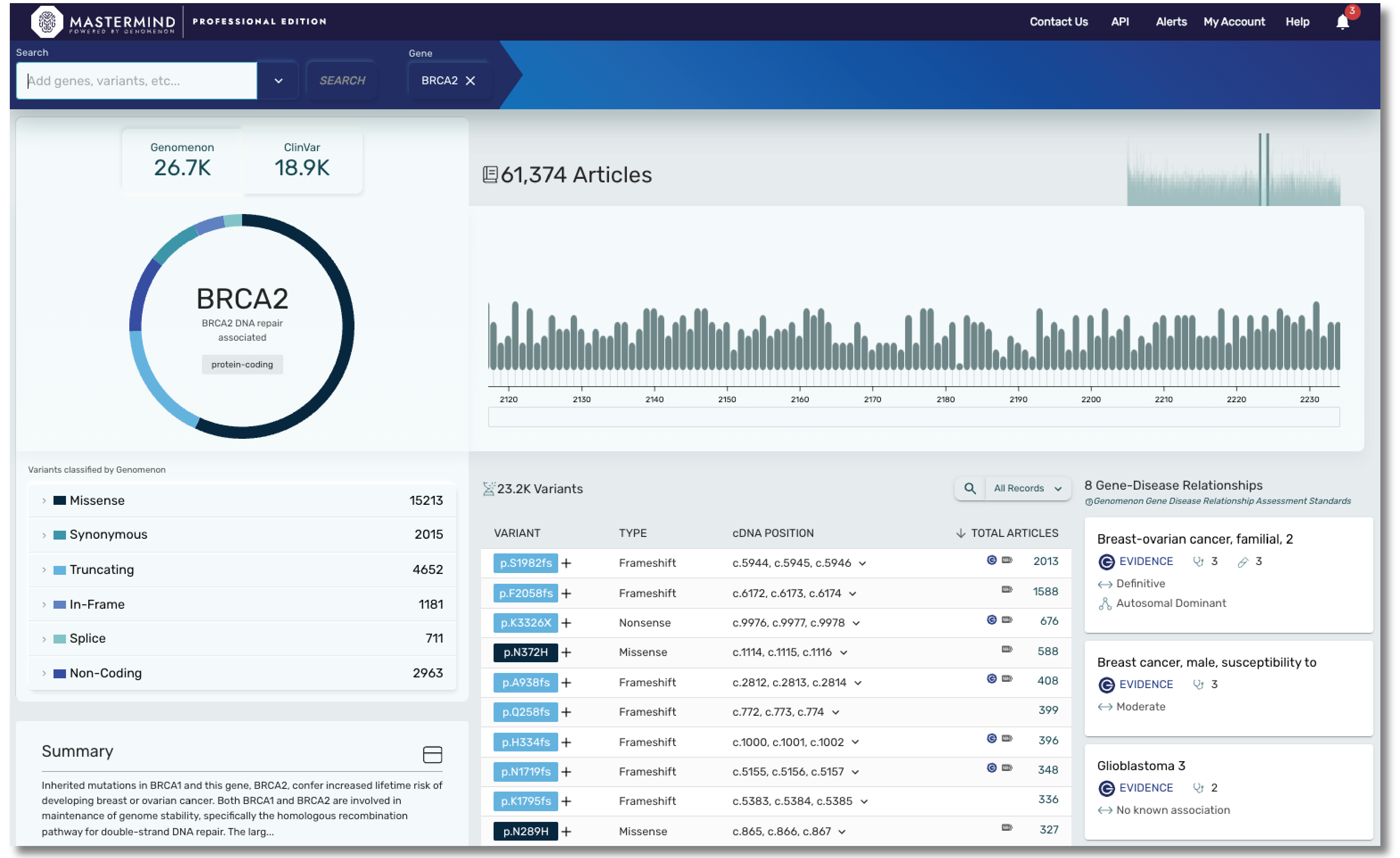
With Mastermind PRO, you can enhance productivity through access to:
- Over 9000 gene disease relationships
- Data on over 19,000 genes and over 27M variants
- Articles for Copy Number Variants
- Curated content covering hereditary and rare disease, including genes found in most cardiac panels, hereditary cancer panels, and much more. Our cardiac panel includes over 140 genes associated with arrhythmias, cardiomyopathies, and syndromic conditions that include cardiology phenotype. Our hereditary cancer panel includes variant level insights on 115 genes, including 16 genes curated by ClinGen.
- Curated content on ACMG secondary evidence, encompassing all 81 genes.
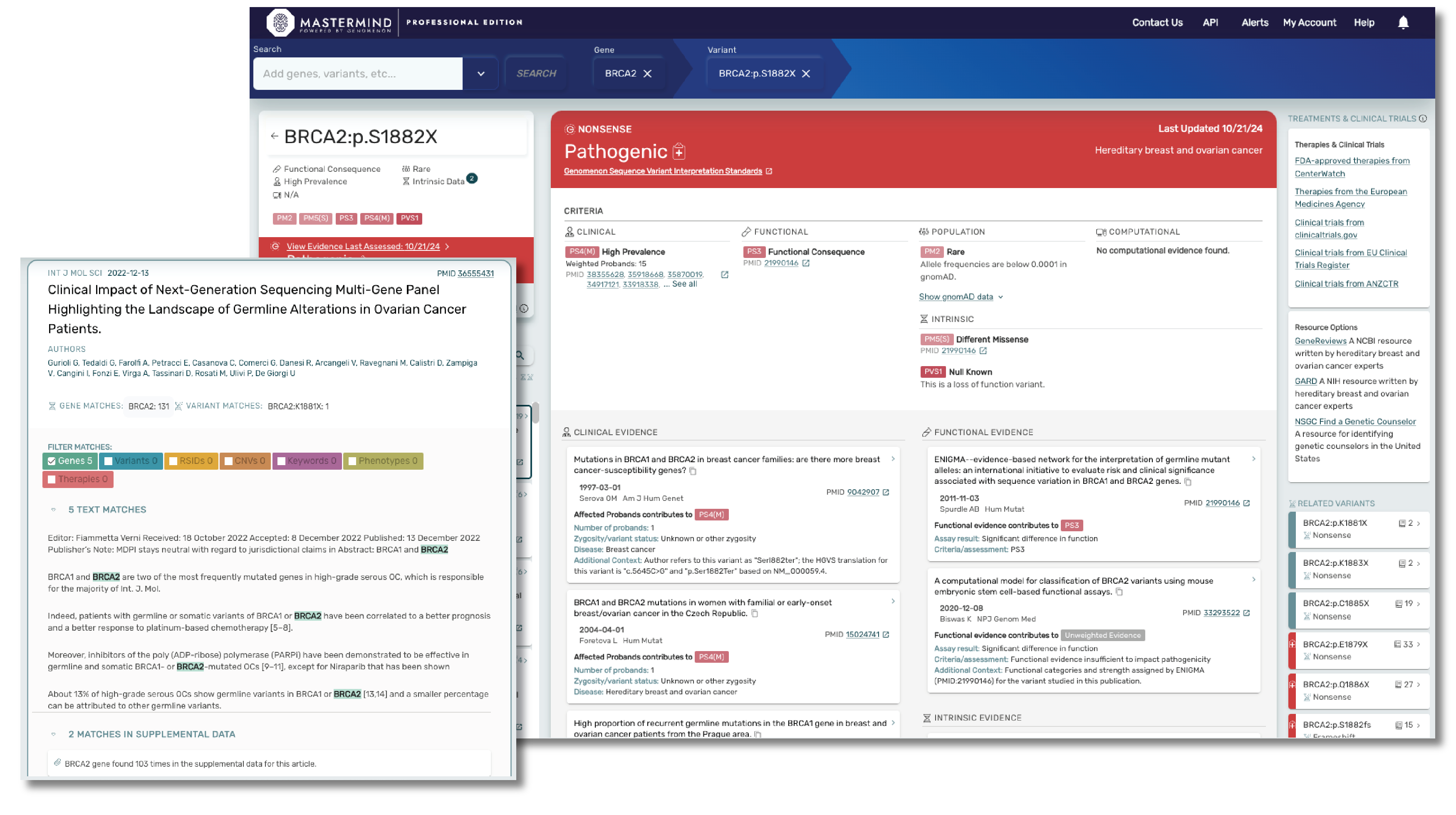
Mastermind CORE is our free version of this platform that enables you to access, understand and leverage the features available in PRO, but for a limited subset of genes. Accessing insights outside of these genes, including indexed information on over 27M variants, 10M full text articles and all of our curated content will require a PRO subscription.
Key Statistics
19,000
genes
27,000,000
variants
10,000,000
full text articles
9,000
gene-disease relationships
3,500,000
supplemental articles
2,000
clinical labs using Mastermind
Mastermind Plans

















Interested in a somatic variant knowledgebase?
CKB is the standard in evidence-based interpretation of complex cancer genomic profiles.
*Stats as of January 2025.
Talk to a Genomenon expert.
